Catalysis and Functional Systems
Catalysts for accelerating water splitting, oxygen evolution reaction (OER) and hydrogen evolution reaction (HER).
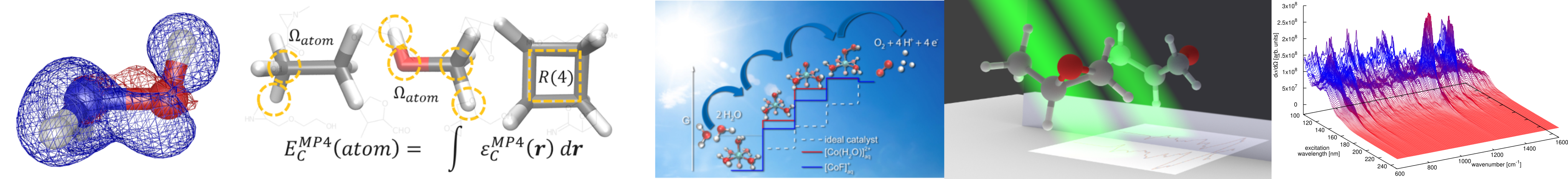
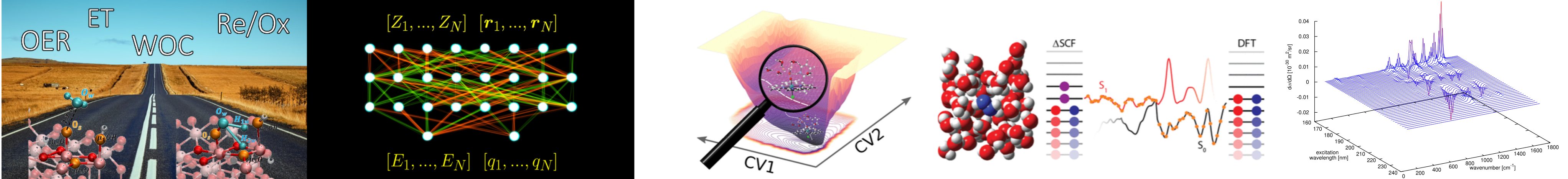
During the past decades, special emphasis has been placed on the development of efficient water oxidation catalysts (WOCs) to overcome the major roadblock in the way of solar hydrogen generation by photosynthesis. A prerequisite for smart design of more efficient WOCs is the understanding of the entire water splitting process (oxygen evolution and hydrogen evolution reactions). The catalyst and its water oxidation behaviour have thus to be elucidated thoroughly. This includes the detailed structure of the catalyst as well as the mechanism of water splitting and related reaction networks. Accordingly, the aim of the present research is to provide, via forefront DFT-MD and metadynamics simulations, an unprecedented understanding of homogeneous catalysts as well as semiconductor catalysts at the interface with explicit solvent. A full understanding of the kinetics, thermodynamics and involved energies behind the water splitting and structural, electronic and mechanical properties is aimed at.
Advancing computational approaches for study of functional systems
We have been interested in smart in silico design of novel catalysts, approaches for calculation of functional properties (e.g. pKa values) via DFT-based molecular dynamics, and highly accurate methods beyond DFT for unprecedented insight into electronic structure of catalysts or functional systems in. general.
Related works:
F. Creazzo, S. Luber
Modeling of solid-liquid interfaces for water splitting catalysis
Book Chapter, Molecular Sciences and Chemical Engineering, Elsevier, 2023
R. Han, S. Luber, G. L. Manni
Magnetic Interactions in a [Co(II)3Er(III)(OR)4] Model Cubane through Forefront Multiconfigurational Methods
J. Chem. Theory Comput. 2023, 19, 10, 2811–2826
N. Plainpan, R. Ketkaew, S. Luber, K. Sivula
Enabling Direct Photoelectrochemical H₂ Production using Alternative Oxidation Reactions on WO₃
Chimia 2023, 77, 110
P. Adams, F. Creazzo, T. Moehl, R. Crockett, P. Zeng, Z. Novotny, S. Luber, W. Yang, S. D. Tilley
Solution phase treatments of Sb2Se3 heterojunction photocathodes for improved water splitting performance
J. Mater. Chem. A, 2023, 11, 8277–8284
L. Schneider, M. Kalt, S. Koch, S. Sithamparanathan, V. Villiger, J. Mattiat, F. Kradolfer, E. Slyshkina, S. Luber, M. Bonmarin, C. Maake, B. Spingler
BODIPY-Based Photothermal Agents with Excellent Phototoxic Indices for Cancer Treatment
J. Am. Chem. Soc. 2023, 145, 8, 4534–4544
R. Ketkaew, F. Creazzo, S. Luber
Closer Look at Inverse Electron Demand Diels–Alder and Nucleophilic Addition Reactions on s-Tetrazines Using Enhanced Sampling Methods
Top Catal, 2022, 65, 366–382
F. Creazzo, R. Ketkaew, S. Luber
Effects of surface wettability on (001)-WO and (100)-WSe: A spin-polarized DFT-MD study
Appl. Surf. Sci. 2022, 601, 154203
N. Weder, N. S. Grundmann, B. Probst, O. Blacque, R. Ketkaew, F. Creazzo, S. Luber, R. Alberto
Two Novel Dinuclear Cobalt Polypyridyl Complexes in Electro- and Photocatalysis for Hydrogen Production: Cooperativity increases Performance
ChemSusChem 2022, e202201049
F. Creazzo, S. Luber
Explicit solvent effects on (1 1 0) ruthenium oxide surface wettability: Structural, electronic and mechanical properties of rutile RuO2 by means of spin-polarized DFT-MD
Applied Surface Science, 2021, 570
F. Creazzo, S. Luber
Water-Assisted Chemical Route Towards the Oxygen Evolution Reaction at the Hydrated (110) Ruthenium Oxide Surface: heterogeneous catalysis via DFT-MD & metadynamics simulations
Chem. Eur. J., 2021, 27,17024-17037
M. Schilling, R. Ketkaew and S. Luber
How ab initio Molecular Dynamics Can Change the Understanding on Transition Metal Catalysed Water Oxidation
Chimia, 2021, 75, 03, 195-201
M. Schilling, R.A. Cunha, S. Luber
Enhanced Ab Initio Molecular Dynamics Exploration Unveils the Complex Role of Different Intramolecular Bases on the Water Nucleophilic Attack Mechanism
ACS Catal., 2020, 10, 14, 7657-7667
R. Han, S. Luber
Complete active space analysis of a reaction pathway: Investigation of the oxygen–oxygen bond formation
J. Comput. Chem., 2020, 41, 1586-1597
M. Schilling, R.A. Cunha, S. Luber
Zooming in on the O–O Bond Formation—An Ab Initio Molecular Dynamics Study Applying Enhanced Sampling Techniques
J. Chem. Theory Comput., 2020, 16, 4
M. Schilling, S. Luber
Determination of pKa values via ab initio molecular dynamics and its application to transition metal-based water oxidation catalysts
Inorganics, 2019, 7 (6), 73
S. Luber
Advancing Computational Approaches for Study and Design in Catalysis
Chimia, 2018, 72 (7-8), 508-513
M. Schilling, M. Böhler, S. Luber
Towards the rational design of the Py5-ligand framework for ruthenium-based water oxidation catalysts
Dalton Trans., 2018, 47, 10480-10490
F. H. Hodel, S. Luber
Dehydrogenation free energy of Co2+(aq) from density functional theory-based molecular dynamics
J. Chem. Theory Comput., 2017, 13, 974–981.
F. H. Hodel, S. Luber
Redox-Inert Cations Enhancing Water Oxidation Activity: The Crucial Role of Flexibility
ACS Catal., 2016, 6, 6750–6761.
M. Schilling, G. R. Patzke, J. Hutter, S. Luber
Computational investigation and design of cobalt aqua complexes for homogeneous water oxidation
J. Phys. Chem. C, 2016, 120, 7966–7975.
F. H. Hodel, S. Luber
What influences the water oxidation activity of a bioinspired molecular CoII4O4 cubane? An in-depth exploration of catalytic pathways
ACS Catal., 2016, 6, 1505–1517.
F. Evangelisti, R. Moré, F. Hodel, S. Luber, G. R. Patzke
3d-4f {CoII3 Ln(OR)4} cubanes as bio-inspired water oxidation catalysts
J. Am. Chem. Soc., 2015, 137, 11076–11084 (highlighted in Chimia Issue 11-2015)
Group members working on this topic:
-
Fabrizio Creazzo
-
Ziwei Chai
-
Rangsiman Ketkaew
-
Deqi Tang