Machine learning / Advancing electronic structure
Molecular properties
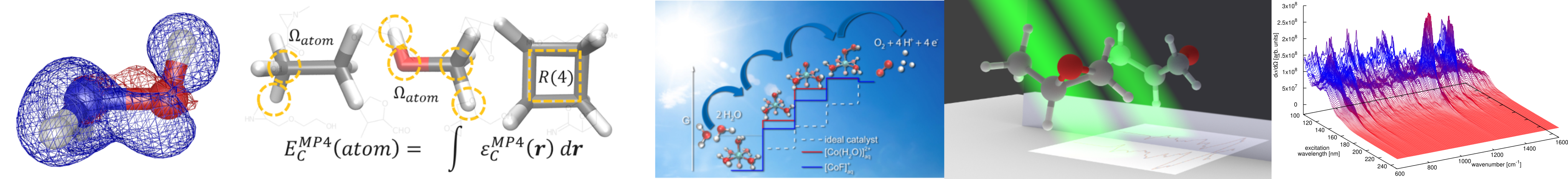
We target on the development of machine learning (ML) approaches for molecular properties (energy, force, etc). Molecular dynamic trajectory-based ML force field is introduced to accelerate ab initio molecular dynamics (AIMD) simulation. Energy density-based descriptors are constructed to predict correlation energy at post-Hartree–Fock level. Among these works, feature engineering is of special interest with the hidden information of chemical systems encoded, so we do not treat everything inside a “black-box”.
Dynamic properties
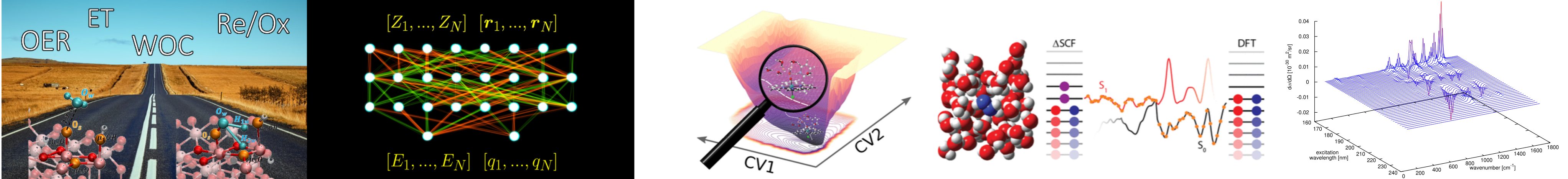
We develop ML algorithms for biasing enhanced sampling methods, e.g., metadynamics. Our current interest focuses on the development of collective variables for complicated chemical system using the deep learning (DL) technique. To improve the learning ability of the DL model, we also work on the development of new structural representations that can be used for arbitrary chemical systems. In addition, an in-house software suite has been developed and used in real world chemistry problems in collaboration with experimentalists.
Related works
R. Han, J. Mattiat, S. Luber
Automatic purpose-driven basis set truncation for time-dependent Hartree–Fock and density-functional theory
Nat. Commun. 2023, 14, 106
R. Ketkaew, F. Creazzo, S. Luber
Machine Learning-Assisted Discovery of Hidden States in Expanded Free Energy Space
J. Phys. Chem. Lett. 2022, 13, 7, 1797–1805
R. Han, R. Ketkaew, S. Luber
A Concise Review on Recent Developments of Machine Learning for the Prediction of Vibrational Spectra
J. Phys. Chem. A 2022, 126, 6, 801-812
R. Han, S. Luber
Fast Estimation of Møller–Plesset Correlation Energies Based on Atomic Contributions
J. Phys. Chem. Lett., 2021, 12, 22, 5324-5331
R. Han, M.Rodríguez-Mayorga, S. Luber
A Machine Learning Approach for MP2 Correlation Energies and Its Application to Organic Compounds
J. Chem. Theory Comput., 2021, 17, 2, 777–790
R. Han, S. Luber
Trajectory-based machine learning method and its application to molecular dynamics
Molecular Physics, 2020, 118:19-20
Group members working on this topic:
- Rangsiman Ketkaew