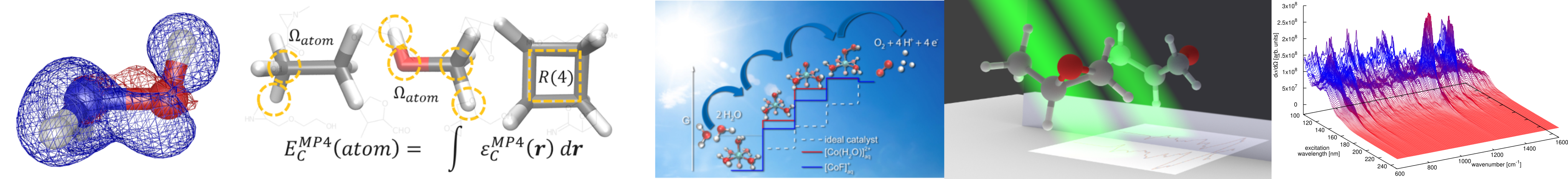
Spectroscopy
A special emphasis is put on theoretical spectroscopy in our group. The calculation and analysis of electronic and vibrational spectra by static and dynamic methods using e.g. perturbation theory, real-time propagation, and subsystem DFT are some of the research directions we have been following. In recent years, we have especially been interested in modelling of spectra for condensed phase systems. Besides studies for the inclusion of solvent effects in static calculations, we have focused on ab initio molecular dynamics as a tool to describe condensed phase systems and their dynamics at ambient conditions. Apart from the use of density functional theory embedding, we have, for instance, presented efficient computational approaches for Raman (optical activity) and sum frequency generation. Other directions have concerned efficient calculation of entire excitation profiles and off-, near- and on-resonance spectra as well as spectroscopic signatures for chiral systems such as electronic circular dichroism.
Related works:
E. Ditler, J. Mattiat, S. Luber
The position operator problem in periodic calculations with an emphasis on theoretical spectroscopy
Phys. Chem. Chem. Phys., 2023, 25, 14672-14685
E. Ditler, C. Kumar, S. Luber
Vibrational circular dichroism spectra of natural products by means of the nuclear velocity perturbation theory
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 298 (2023) 122769
R. Han, J. Mattiat, S. Luber
Automatic purpose-driven basis set truncation for time-dependent Hartree–Fock and density-functional theory
Nat. Commun. 2023, 14, 106
A. Kelemen, S.Luber
On the vibrations of formic acid predicted from first principles
Phys. Chem. Chem. Phys., 2022, 24, 28109-28120
J. Mattiat, S. Luber
Comparison of Length, Velocity, and Symmetric Gauges for the Calculation of Absorption and Electric Circular Dichroism Spectra with Real-Time Time-Dependent Density Functional Theory
J. Chem. Theory Comput. 2022, 18, 9, 5513–5526
E. Ditler, T. Zimmermann, C. Kumar, S. Luber
Implementation of Nuclear Velocity Perturbation and Magnetic Field Perturbation Theory in CP2K and Their Application to Vibrational Circular Dichroism
J. Chem. Theory Comput. 2022, 18, 4, 2448-2461
E. Ditler, S. Luber
Vibrational spectroscopy by means of first-principles molecular dynamics simulations
WIREs Comput Mol Sci. 2022;e1605
J. Mattiat, S. Luber
Recent Progress in the Simulation of Chiral Systems with Real Time Propagation Methods
Helv. Chim. Acta 2021, 104, e2100154
L. Schreder, S. Luber
Local approaches for electric dipole moments in periodic systems and their application to real-time time-dependent density functional theory
J. Chem. Phys., 2021, 155, 134116
E. Ditler, C. Kumar, and S.Luber
Analytic calculation and analysis of atomic polar tensors for molecules and materials using the Gaussian and plane waves approach
J. Chem. Phys., 2021, 154, 104121
J. Mattiat, S. Luber
Time Domain Simulation of (Resonance) Raman Spectra of Liquids in the Short Time Approximation
J. Chem. Theory Comput., 2021, 17, 1, 344-356
J. Mattiat, S. Luber
Vibrational (resonance) Raman optical activity with real time time dependent density functional theory
J. Chem. Phys., 2019, 151, 234110,
J. Mattiat, S. Luber
Electronic circular dichroism with real time time dependent density functional theory: Propagator formalism and gauge dependence
Chem. Phys., 2019, 527 (1 November 2019), 110464
S. Luber
Localized molecular orbitals for calculation and analysis of vibrational Raman optical activity
Phys. Chem. Chem. Phys., 2018, 20, 28751-28758
J. Mattiat and S. Luber
Efficient calculation of (resonance) Raman spectra and excitation profiles with real-time propagation
J. Chem. Phys., 2018, 194, 174108
S. Luber
Raman optical activity spectra from density functional perturbation theory and density functional theory-based molecular dynamics
J. Chem. Theory Comput., 2017, 13, 1254–1262.
S. Luber
Sum frequency generation of acetonitrile on rutile (110) surface from density functional theory-based molecular dynamics
J. Phys. Chem. Lett., 2016, 7, 5183–5187.
P. Oulevey, S. Luber, B. Varnholt, T. Bürgi
Symmetry Breaking in Chiral Ionic Liquids Evidenced by Vibrational Optical Activity
Angew. Chem. Int. Ed., 2016, 55, 11787–1790.
S. Luber
Exploring Raman optical activity for transition metals: From coordination compounds to solids
Biomed. Spectrosc. Imaging, 2015, 4, 255–268 (invited review)
S. Luber
Local electric dipole moments for periodic systems via density functional theory embedding
J. Chem. Phys., 2014, 141, 234110.
Group members working on this topic:
-
Johann Mattiat
-
Ravi Kumar
-
Lukas Schreder
-
Anna Kelemen